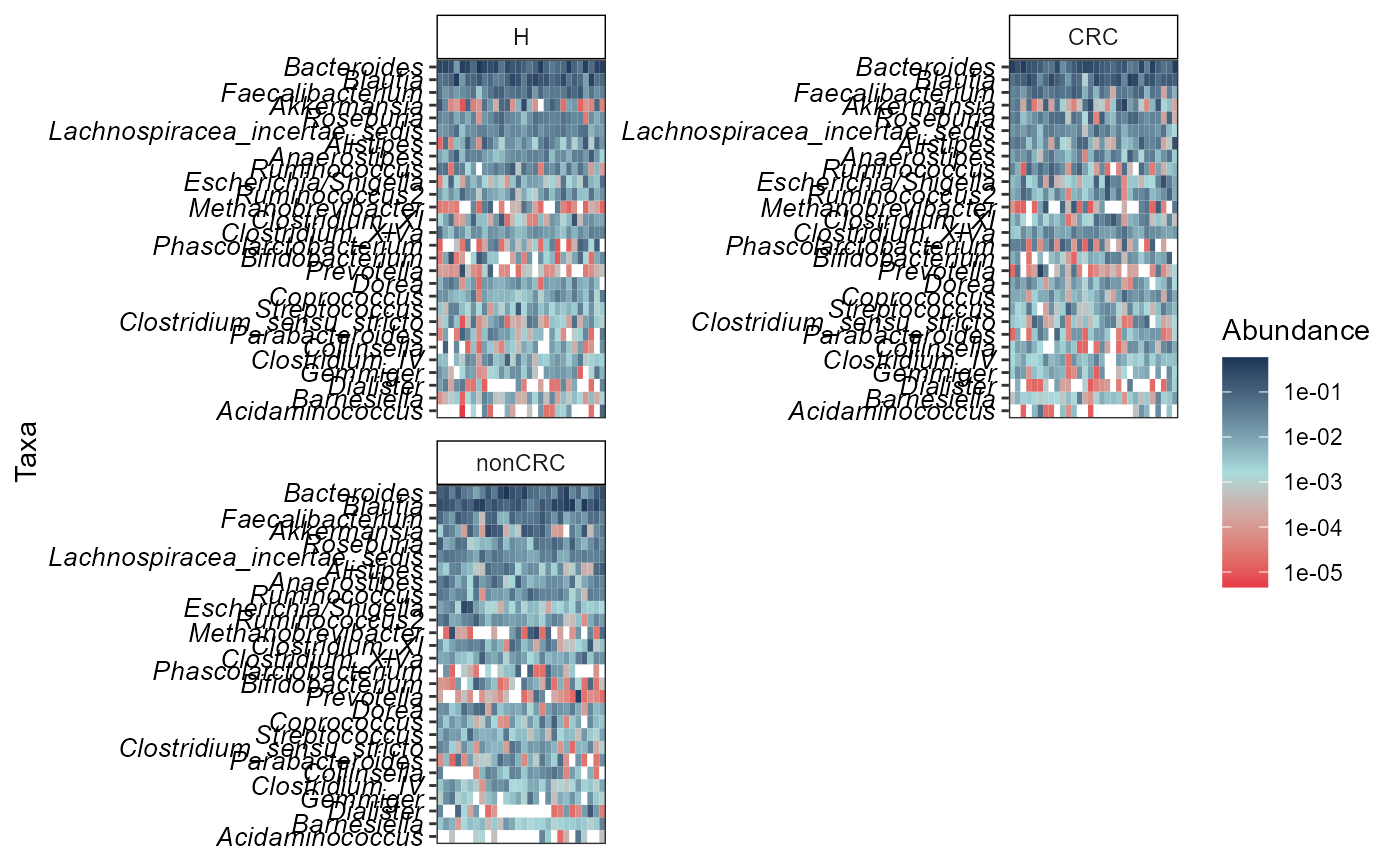
Create a simple heatmap with ggplot2 package.
simple_heatmap(
x,
group.facet = "DiseaseState",
group.order = c("H", "CRC", "nonCRC"),
abund.thres = 0.01,
prev.thres = 0.1,
level = "Genus",
scale.color = "log10",
na.fill = "white",
color.fill = NULL,
taxa.arrange = TRUE,
panel.arrange = NULL,
remove.other = TRUE,
ncol = NULL,
nrow = NULL
)Arguments
- x
phyloseq-classobject.- group.facet
Variable to make facet/panel the plot.
- group.order
Default is NULL. a list specifing order of x-axis. E.g. c("H","CRC","nonCRC")
- abund.thres
= 0.01 check
microbiomepackage aggregate_rare function.- prev.thres
= 0.1 check
microbiomepackage aggregate_rare function.- level
= "Genus" Taxonomic level. OTU/ASV level not supported. Check plot_taxa_heatmap
- scale.color
Scale the tiles colors "log10" or "sqrt"
- na.fill
Color to fill NAs. e.g. "white"
- color.fill
User specified color vectors.
- taxa.arrange
Arrange the order of taxa. User can supply a list of vectors.
- panel.arrange
panels "grid" or "wrap" ggplot's facet_XXX
- remove.other
Rare clubbed as Other to be removed. Logical TRUE/FALSE.
- ncol
if wrap, specify number of columns.
- nrow
if wrap, specify number of rows.
Value
ggplot object.
Details
Wrapper converts phyloseq-class object to long data frame
and generates a heatmap.
Examples
library(microbiome)
library(microbiomeutilities)
library(dplyr)
data("zackular2014")
p0 <- zackular2014
p0.rel <- transform(p0, "compositional")
p <- simple_heatmap(p0.rel,
group.facet = "DiseaseState",
group.order = c("H", "CRC", "nonCRC"),
abund.thres = 0.01,
prev.thres = 0.1,
level = "Genus",
scale.color = "log10",
na.fill = "white",
color.fill = NULL,
taxa.arrange = TRUE,
remove.other = TRUE,
panel.arrange = "wrap",
ncol = 2,
nrow = 2
)
#> An additonal column Sam_rep with sample names is created for reference purpose
print(p)
#> Warning: Transformation introduced infinite values in discrete y-axis